Accurate and Highly Accurate
datasets of protein structure models
General
Description of the dataset
This database contains a
total of 55 highly accurate and 57 accurate protein structure models. Both sets
were obtained after a careful classification and selection of models from a
large database composed of 3.375 “good models”. These “good models” were
calculated by large-scale comparative modelling of the protein chains
representative of the Protein Data Bank (PDB, Berman et al., 2002) and have
been used in previous studies (Sánchez and Sali, 1998; Melo et al., 2002; Melo and
Marti-Renom, 2006). Accurate and highly accurate sets of models were derived to
assess the performance of knowledge-based potentials in the detection of
structural errors of small magnitude (see Figure 1). The first set (class A)
contains 55 highly-accurate protein models. The second set (class B) contains
57 accurate protein models. The highly-accurate protein models from the class A
set have more than 95% equivalent a-carbons with their
corresponding native structures and a total root mean square deviation (RMSD)
of less than 1.1 Å for all a-carbons. The
accurate protein models from the class B set have more than 90% equivalent a-carbons with their corresponding native structures and a total RMSD of
more than 1.5 Å for all a-carbons.
Building the
sets
The 57 accurate
and 55 highly-accurate protein models were selected from a existing set of
3,375 models with a correct fold that has been described previously (Sanchez
and Sali 1998; Melo et al. 2002). This original set of 3,375 models was built
by the comparative modelling of representative chains of the Protein Data Bank
(PDB) (Berman et al. 2002). The models were built based on the correct
templates and mostly correct alignments between the target sequences and the
template structures. The models were obtained by applying MODPIPE to 1.085
representative chains of the PDB (Sanchez and Sali 1998). These representative
sequences corresponded to the protein chains in PDB that shared <30% sequence
identity or were >30 residues different in size. The templates for
comparative modelling, selected by MODPIPE, were 1,637 PDB chains with <80%
sequence identity to each other or more than 30 residue difference in length.
Each target sequence was aligned separately with each one of the 1,637 known
structures using the program ALIGN that implements local sequence alignment by
dynamic programming (Altschul 1998). Only the target-template alignments with a
significance score higher than 22 nats (corresponding approximately to the
PSI-BLAST e-value of 10−4) were used,
resulting in 3,993 models. Models with <30% structural overlap with the
actual experimentally determined structure were eliminated. Structural overlap
was defined as the fraction of the equivalent Cα atoms upon least
squares superposition of the two structures with the 3.5 Å cut-off. This
procedure also removed models based on correct templates that had a poor
alignment and models based on templates that had large domain or rigid body
movements with respect to the target structure. The final set contained 3,375
correct models (Melo et al. 2002). These models are available here.
The set of 3,375
correct models was initially filtered by updating the target and template
structures currently available at the PDB and checking the sequence alignments
originally used to build the models. A total of 132 models presented
inconsistencies between the target sequence in the original alignment used to
build the model and the current target sequence available at the PDB. These 132
models were removed, thus resulting in a total of 3,243 entries (see Figure 1).
Then, a second filter was applied and we selected only those protein models of
a length larger than 100 residues for which more than 90% of their residues was
possible to model. Finally, as explained above, two independent filters were
applied to select those models belonging to the class A and class B sets (see Figure
1). All models in both sets were built for target monomeric proteins. PDB files
of each model superposed with its corresponding native structure can be
downloaded from the links available in the table below.
Identification
and definition of those residues incorrectly modeled
To
identify those residues that contained structural errors, all accurate and
highly-accurate protein models were optimally superposed to their corresponding
native target structures using MODELLER software release 7v7 (Sali et al. 2001).
Those residues that had an RMSD larger than 1.8 Å for all main chain atoms and a total sidechain RMSD
larger than 3.5 Å were defined as wrongly
modeled. Otherwise, the residues were defined as correctly
modeled. This binary classification of structural quality for each residue is
called ‘real classification’. Upon this classification scheme, 201 residues
(1.95%) are defined as wrongly modeled in the 55 models belonging to the class
A set, which contains a total of 10,295 residues. In the 57 models belonging to
the class B set, 1,257 residues (11.73%) are defined as wrongly modeled, from a
total of 10,714 residues. Though arbitrary, the definition of residues wrongly
modeled upon these RMSD cutoffs was based on the visual inspection of all
protein models after optimal superposition with their corresponding native
structures. This definition of error clearly correlates with the observed
structural deviation. All protein models, along with their corresponding error
definitions and computer scripts for RASMOL software (Sayle and Milner-White
1995; Bernstein 2000) to visualize them graphically in three-dimensions can be
downloaded from the table below.
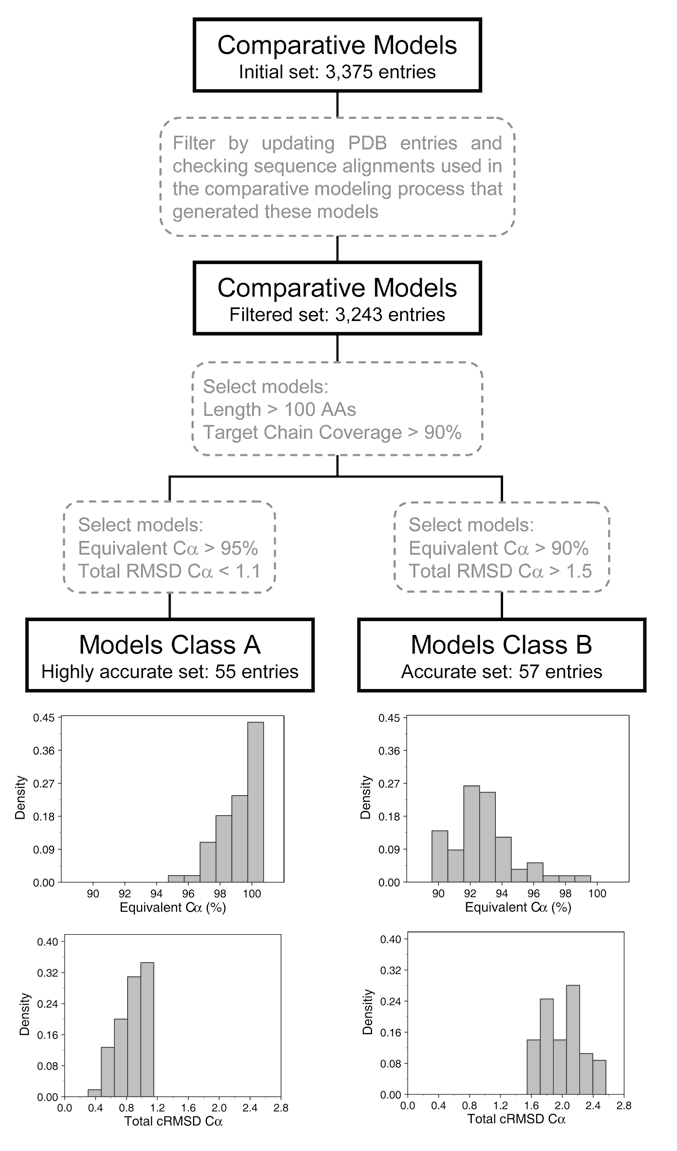
Figure 1: Flow
chart of the procedure used to build the sets of comparative protein models. The
source protein model set was originally constituted of 3.375 models with the
correct fold, which were built in a previous study using Modpipe (Sanchez and Sali 1998). After
applying several filtering criteria two subsets of models were produced.
Raw data
|
File name |
Type |
File format |
Number of elements |
General description |
|
Models class A |
||||
|
list |
plain text |
55 model names |
A list of class
A models |
|
|
coordinates |
PDB format
(plain text) |
55 files |
The PDB files
of the models (“mdl”) class A. |
|
|
coordinates |
PDB format
(plain text) |
55 files |
The PDB files
of each model class A superposed (“spd”) with their corresponding native
structure. |
|
|
data file |
plain text |
55 files |
Four column
file. 1) residue number, 2) backbone cRMSD, 3) sidechain cRMSD, 4)
incorrectly modelled residue (1) or correctly modelled residue (0). |
|
|
RasMol script |
plain text |
55 files |
RasMol scripts
to visualize those residues wrongly modelled. |
|
|
Models class B |
||||
|
list |
plain text |
57 model names |
A list of class
B models |
|
|
coordinates |
PDB format
(plain text) |
57 files |
The PDB files
of all models (“mdl”) class B. |
|
|
coordinates |
PDB format
(plain text) |
57 files |
The PDB files
of each model class B superposed (“spd”) with their corresponding native
structure. |
|
|
data file |
plain text |
57 files |
Four column
file. 1) residue number, 2) backbone cRMSD, 3) sidechain cRMSD, 4)
incorrectly modelled residue (1) or correctly modelled residue (0). |
|
|
RasMol script |
plain text |
57 files |
RasMol scripts
to visualize those residues wrongly modelled. |
|
References
|
Altschul, S. (1998) |
|
Berman, H.M., Battistuz, T., Bhat, T.N., Bluhm,
W.F., Bourne, P.E., Burkhardt, K., Feng, Z., Gilliland, G.L., Iype, L., Jain,
S., Fagan, P., Marvin, J., Padilla, D., Ravichandran, V., Schneider, B.,
Thanki, N., Weissig, H., Westbrook, J.D., and Zardecki, C. (2002) |
|
Melo, F. and Marti-Renom, M. (2006) |
|
Melo, F., Sánchez, R., and Sali, A. (2002) |
|
Sali, A., Fiser, A.,
Sanchez, R., Marti-Renom, M.A., Jerkovic, B., Badretdinov, A., Melo, F.,
Overington, J., and Feyfant, E. (2001). MODELLER,
A Protein Structure Modeling Program, Release 6v0. |
|
Sánchez R. and Sali, A. (1998) |
|
Sayle, R., and
Milner-White, E.J. (1995). RasMol: Biomolecular graphics for all. TIBS 20:
374. |